Call for Master and Undergraduates Students: Join My Bioinformatics Research Group!
I am currently seeking motivated and dedicated Master’s students (research track preferred) and undergraduates in Biomedical Sciences to join my bioinformatics research projects.
Due to the intensive nature of the work, I cannot accept students with less than three semesters remaining (example: if you plan to graduate in Fall 2026, you need to join my group at least in Fall 2025). All projects are open to both Master and Undergraduate students.
1. Improving precision medicine efforts by investigating the evolution of obesity in the Qatari population through the study of ancient human genomes
In recent years, analysis of ancient DNA genomes has facilitated the detection of pathogens that caused historical morbidity and mortality events. However, to date, there have been no such studies done on Arabs populations living within the Middle Eastern and Northern African (MENA) region. In our previous work in the Qatar Genome Project (QGP), we showed how the Qatari population can represent the genetic structure of over 400 million individuals from across the MENA region, most of whom are understudied. By linking the ancient DNA to the modern MENA population and studying the evolution of pathogenic variants, it could help us to understand the nature of diseases.
The aim is to understand how changes in the genomic sequence over time shaped present-day predisposition to diseases. This comparison will provide an overview of the pathogenicity of SNPs and their linkage with diseases in the Middle East over thousands of years. Furthermore, we also aim to describe the movement of disease in line with the spread of Arab genetic ancestries throughout the MENA region. The information gathered by studying the ancient DNA of Middle Eastern individuals will enhance our understanding of pathogenic emergence and disease evolution over time across the MENA region and assist in the development of more personalized and precise treatment and prevention strategies.
2. Application of whole genome sequencing (WGS) to uncover insights to the relationship between historical migration of Malay population and genetic disease
In our previous study (Razali et al., 2021), we hypothesize that Peninsular Arabia was the launching pad for humanity to venture to other parts of the world after the migration out of Africa. However, the gene flow to the Southeast Asia part of the world was not represented. Like Arabia, the Southeast Asia region is under-represented in genomics. Archaeological evidence showed that modern humans have occupied Southeast Asia for 60kya, far longer than even the settlement in East Asia (estimated 40kya). Current evidence suggests that SEA was occupied by Hòabìnhian hunter-gatherers until the Neolithic period, when farming economies expanded, restricting foraging groups to remote habitats (McColl et al., 2018). It has been previously shown that the ancestors of the modern-day Negritos are the first settlers in the Malay Archipelago and subsequently interbreed with populations coming from the South and East Asian regions (Yew et al., 2018). In recent years, many studies have uncovered genetic data of ancient humans worldwide. This genetic data provides opportunities for researchers to compare them against the modern population.

In our study, we aim to examine the genomic diversity between the different modern Malays populations with ancient genomes and other modern world populations. To achieve this, we plan to do the following (a) To identify the presence/absence of genetic variants of interest between modern and ancient genomes (b)To predict the population structure of the Malay-subgroups (c) To describe the gene flow between different Malay sub-ethnic groups with other modern and ancient ancestries (d)To identify the demographic history of the Malay population through effective population size.
The resulting findings will yield practical benefits for public health and will directly align with the national research strategy and vision. As a result, it will enhance our understanding of pathogenic emergence, disease evolution and disease predisposition across the Southeast Asia region. Our methodology represents an innovative approach that leverages cutting-edge sequencing technologies to explore clinical diagnostic inquiries.
3. Exploring Admixture-Driven Somatic and Germline Mutations in Breast Cancer among the Qatari Population
The aim of this project is to study the somatic mutation signature profile in Qatari populations using genetic ancestry. Such signature profiles in our target dataset could be exploited as novel/early biomarkers and/or prospective treatment targets to manage human cancer patients, including breast cancer.

To achieve this aim, we present 3 objectives. The first is to perform mutational analysis in the tumour and link its status with therapy response in Qatari patients. The second is to assess gene expression in tumor tissue this to create predictor of response to specific therapy. The third is to track the pathogenic genetic variants across different historical periods using ancient DNA from the Middle East and Northern African (MENA) region. We plan to perform whole genome sequencing (WGS) and whole transcriptome sequencing (WTS) of 200 breast cancer patients. For WGS, we will analyze the samples from tumour and match normal tissues. In addition, the match normal tissues will be also used to predict germline mutations. We will use this germline mutations together with other germline resources to identify and cluster our 200 patients into genetic ancestries. This would allow us to treat the 200 patients as heterogenous groups rather than one homogenous cohort. We will then compare between inter and intra ancestry groups and with other world population to identify somatic signature profile.
Additionally, we will study the germline mutation profile of our patient cohort with published ancient DNA from around the MENA region by tracking the presence and/or absence of mutations over thousands of years. This will allow us to identify the evolutionary mechanism that gave rise to the germline mutations associated with breast cancer. In terms of the expected outcome and impact from this study it can be seen from the perspective of the United Nation Sustainable Development Goals (SDGs) such as in promoting health and well-being (SDG 3). By having a somatic mutation profile signature based on genetic ancestry, it can improve therapeutic efficacy while decreasing the toxicity of specific therapies since it will provide insights into unique signature markers at the subpopulation level. This is critical in improving precision medicine initiatives in Qatar since it enables the development of personalized treatment strategies that consider the population’s distinct genetic attributes. Finally, this project will raise awareness of somatic mutations because it is the first large-scale somatic study in Qatar. It has the potential to be an excellent training experience for Qatar University staff and students and will help in producing a new generation of researchers capable of performing complex cancer research thus paving the way for establishing knowledge-economy workers in Qatar.
4. Historical migration of key wildlife in Qatar and their adaptation to extreme climate through the study of ancient and modern DNA.
Qatar, a nation characterized by its harsh desert climate and saline marine environments, has long been home to a unique array of wildlife species that have adapted to these extreme conditions. Understanding the historical migration patterns and subsequent genetic adaptations of these species is crucial for conserving biodiversity and ensuring ecosystem resilience in the face of ongoing climate change. Ancient DNA (aDNA) analysis, combined with the study of modern genomes, offers a powerful approach to uncovering the evolutionary processes that have enabled certain species to thrive in such challenging environments. This study aims to investigate the genetic mechanisms underlying the adaptation of key wildlife species in Qatar to extreme climatic conditions by comparing their ancient and modern genomes. Through comprehensive genome assembly, annotation, and functional analysis, the study will provide insights into how these species have evolved over time and adapted to the high temperatures and salinity of the region.

The primary aim of this study is to elucidate the genetic adaptations of key wildlife species in Qatar to extreme climatic conditions by comparing ancient and modern DNA. Specifically, the study seeks to identify the genetic factors that have enabled these species to survive and thrive in hot, saline environments and to compare these adaptations with those of the same species in colder, temperate climates. We will perform high-quality genome assembly and annotation for ancient and modern DNA samples of selected wildlife species in Qatar. Identify and annotate functional elements in the genomes, such as genes, regulatory regions, and non-coding RNAs, that are involved in adaptation to extreme climates. Compare the genomes of the same species from Qatar (hot, saline environment) with those from colder, temperate climates (non-saline environment) to identify genetic differences associated with environmental adaptation. Investigate the evolutionary processes, such as natural selection and genetic drift, that have shaped the genetic adaptations of these species to Qatar’s extreme climate. Reconstruct the historical migration routes of these species using aDNA and assess how these movements have influenced genetic diversity and adaptation.
This study will significantly enhance our understanding of the genetic basis of adaptation to extreme environments, particularly in the context of climate change. By uncovering the evolutionary history and genetic adaptations of key wildlife species in Qatar, the research will contribute to the conservation of biodiversity in the region and inform strategies for managing and protecting wildlife in other arid and saline environments. Additionally, the insights gained from comparing ancient and modern genomes will provide valuable information on how species may continue to adapt to changing climates in the future, offering a predictive framework for assessing the resilience of ecosystems globally.
5. Development of bioinformatics pipeline (Highly recommended for Capstone Master students)
The rapid advancements in high-throughput sequencing technologies have revolutionized the field of bioinformatics, enabling the analysis of vast genetic datasets. While Genome-Wide Association Studies (GWAS), Polygenic Risk Scores (PRS), and RNA sequencing (RNA-seq) remain essential tools for understanding the genetic basis of complex diseases and traits, the growing importance of microbiome and metagenome analysis cannot be overlooked. Microbiome and metagenome data offer valuable insights into the role of microbial communities in human health and disease, and the development of efficient bioinformatics pipelines for analyzing this type of data is crucial.
This Master’s project aims to expand upon existing bioinformatics pipelines to include microbiome and metagenome analysis, providing a comprehensive suite of tools for researchers. The pipelines will be designed to be user-friendly, scalable, and adaptable to various research needs, facilitating the efficient processing and interpretation of complex genetic, transcriptomic, and microbial data.
The primary aim of this Master’s project is to develop and implement comprehensive bioinformatics pipelines for conducting Genome-Wide Association Studies (GWAS) combined with Polygenic Risk Scores (PRS) analysis, an RNA sequencing (RNA-seq) analysis pipeline and a microbiome/metagenome analysis pipeline. These pipelines will be designed to be user-friendly, scalable, and adaptable to various research needs, facilitating the efficient processing and interpretation of complex genetic and transcriptomic data. The objectives will be to develop the respective pipeline, optimize them, validate or benchmark the result and create user interface/documentation/tutorial.
The development of these bioinformatics pipelines will have a significant impact on the field of biomedical research. By providing researchers with efficient, scalable, and user-friendly tools for GWAS+PRS, RNA-seq analysis, microbiome/metagenome analysis, this project will facilitate the discovery of genetic variants associated with complex diseases and traits, elucidation of gene expression patterns underlying various biological processes and the characterization of microbial communities and their interactions with their hosts. The pipelines will enable researchers at Qatar University to make more informed and accurate interpretations of genetic and transcriptomic data, advancing our understanding of human health and disease.
6. Genetic and transcriptomics characterization of idiopathic calcium oxalate urolithiasis.
Idiopathic calcium oxalate urolithiasis (ICOU) is a common urinary tract disorder characterized by the formation of calcium oxalate stones in the urinary tract. Despite extensive research, the exact etiology and pathogenesis of ICOU remain poorly understood. This study aims to elucidate the genetic and transcriptomic underpinnings of ICOU, potentially leading to novel diagnostic and therapeutic strategies.
The aim is to characterize the genetic landscape of ICOU using whole-genome sequencing, analyze RNA sequencing data to identify differentially expressed genes in ICOU patients compared to controls, integrate genetic and transcriptomic data to identify gene-expression regulatory networks associated with ICOU and discover novel biomarkers for ICOU.
This study has the potential to significantly advance our understanding of the genetic and molecular mechanisms underlying ICOU. The findings may identify novel genetic risk factors for ICOU, leading to improved risk stratification and early intervention, discover new therapeutic targets for ICOU, paving the way for the development of more effective treatments, and provide valuable insights into the pathogenesis of ICOU, contributing to a better understanding of this common urinary tract disorder.
7. Multiomics integration for identifying novel biomarkers and therapeutic targets in hypertension and cardiovascular diseases.
Cardiovascular diseases, such as hypertension, represent a leading cause of morbidity and mortality worldwide, with complex genetic and molecular underpinnings that remain incompletely understood. This comprehensive study leverages the power of multiomics integration to uncover novel insights into the pathogenesis of primary hypertension through the systematic analysis of genomic, proteomic, and metabolomic data from the Qatar Genome Project (QGP) cohort.
Our research integrates data from over 14,000 individuals, applying rigorous inclusion and exclusion criteria based on the 2017 ACC/AHA hypertension guidelines to define normal, elevated, stage 1, and stage 2 hypertension categories. By systematically removing confounding factors such as dyslipidemia, diabetes, chronic kidney disease, and obesity, we aim to identify biomarkers specifically associated with essential hypertension. Building upon previous findings, this study seeks to validate these discoveries and identify novel biomarkers through comprehensive genome-wide association studies (GWAS), proteome-wide association studies (pWAS), metabolome-wide association studies (mWAS), and quantitative trait loci (QTL) analyses.
The integration of machine learning and artificial intelligence approaches enables the identification of complex interaction patterns across multiple omics layers, facilitating the discovery of novel pathogenic pathways and potential therapeutic targets. This multiomics framework provides unprecedented resolution in understanding the molecular mechanisms underlying cardiovascular disease susceptibility in the Middle Eastern population, offering opportunities for precision medicine applications and personalized treatment strategies.
The findings from this study will significantly advance our understanding of pathogenesis, contribute to the development of population-specific biomarker panels for early detection and risk stratification, and inform the design of targeted therapeutic interventions. Furthermore, the methodological framework developed through this research will serve as a foundation for investigating other complex cardiovascular diseases, establishing a comprehensive pipeline for multiomics analysis in cardiovascular health research.
8. Therapeutic Lead Discovery for Metabolic Pathway Disorders.
Our research focuses on discovering new treatments for genetic disorders in the metabolic pathway, a set of biochemical processes that are especially important in Qatar due to high prevalence of related diseases such as classical homocystinuria. These disorders can cause serious neurological and developmental problems, yet current therapies are limited.

Using Qatar Biobank’s unique multi-omics dataset, we integrate computational biology, structural bioinformatics, and drug discovery to identify critical weak points in the methionine pathway. By combining AI-driven drug screening with experimental validation (including zebrafish models), our team aims to discover novel therapeutic compounds that could restore balance to this pathway and improve patient outcomes.
This project not only advances precision medicine in Qatar, but also builds local expertise in next-generation bioinformatics, molecular modeling, and computational drug discovery. Students joining the team will gain hands-on experience with big data analysis, protein structure modeling, AI in drug discovery, and lab validation techniques — preparing them for impactful careers at the intersection of genomics, medicine, and biotechnology.
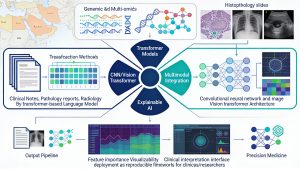
9. Machine Learning Experiments Involving Biomedical Text and Imaging Files
Recent advances in machine learning (ML) and artificial intelligence (AI) have transformed how we analyze complex biomedical data, especially free‑text clinical records and high‑dimensional medical images. However, most existing models are developed on Western cohorts and are not tailored to the genetic, environmental, and clinical characteristics of Middle Eastern populations. This project aims to design, implement, and benchmark ML workflows that integrate biomedical text (such as clinical notes, pathology reports, and radiology narratives) with imaging data (including histopathology slides and radiological scans) to improve disease detection, prognosis prediction, and treatment response modeling